



GMP nel settore farmaceutico: Come individuare i difetti – Prima parte
Quando si parla di GMP (Good manufacturing practices o Norme di buona fabbricazione) in ambito farmaceutico si intende quell’insieme di regole, procedure e linee guida in base alle quali deve essere organizzata la produzione e il confezionamento di farmaci al fine di assicurare la qualità del prodotto, la sicurezza del paziente finale e l’integrità dei dati critici. L’attività di validazione, che svolgiamo noi di SPAI, serve a dimostrare l’impiego di processi che seguono tali criteri.
A prescindere dal metodo analitico impiegato per effettuare l’analisi e valutazione dei rischi di un processo in ambito farmaceutico (ad esempio FTA o FMEA), è necessario avere ben chiari in mente quali siano i difetti che vogliamo evitare e che possono verificarsi durante la produzione e il confezionamento di farmaci. Li abbiamo raggruppati in quattro macroaree:
- Difetti riguardanti le informazioni critiche sulla confezione.
- Difetti riguardanti il farmaco.
- Difetti riguardanti la confezione.
- Difetti riguardanti i dati variabili.
In questa prima parte del nostro viaggio tra i possibili difetti andiamo a vedere quali problematiche possono verificarsi nelle informazioni critiche stampate sulla confezione e nel farmaco stesso.
Difetti riguardanti le informazioni critiche della confezione
Quando si parla di informazioni critiche si intendono i dati già presenti sulla confezione prima che essa venga inserita all’interno del processo di confezionamento, ad esempio:
- Il nome del farmaco.
- Il nome ed eventuale logo della Casa farmaceutica.
- Le informazioni riguardanti il farmaco.
- Le posologie.
- La quantità di prodotto nella confezione.
- Le avvertenze principali (tenere lontano dalla portata dei bambini…)
Quali difetti di questo tipo si possono verificare?
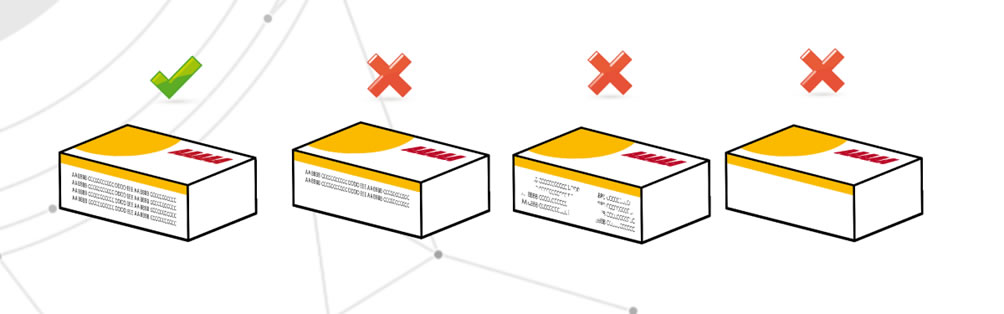
Mancanza di informazioni critiche
In questo caso si intende l’assenza totale o parziale di tali dati, ma anche l’illeggibilità di essi. I problemi, ad esempio, possono derivare da processi di stampa tipografica. Il rischio è quello di non fornire al paziente informazioni complete.
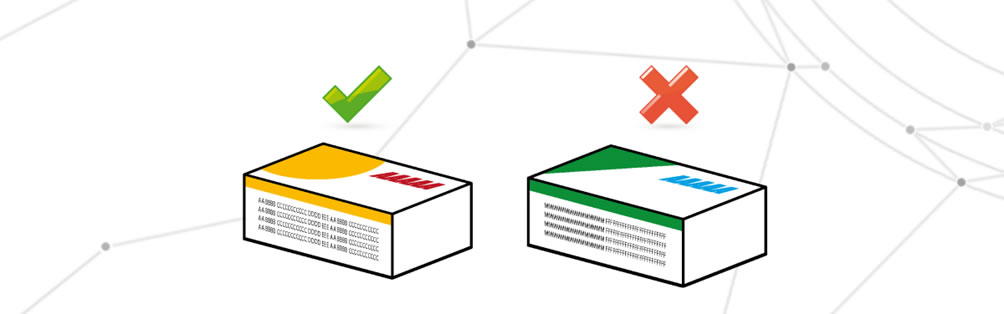
Informazioni critiche non corrispondenti a quelle previste
In questo caso sulla confezione troviamo tutte le informazioni richieste, ma errata è la corrispondenza con il farmaco interno o la correttezza delle stesse informazioni. Ci si può trovare davanti all’utilizzo di dati obsoleti o all’utilizzo di una confezione errata, di norma la causa di questo difetto è il frammischiamento delle confezioni o una non corretta gestione dei magazzini.
Difetti riguardanti il farmaco
In questo caso facciamo riferimento al farmaco vero e proprio, i difetti possono essere provocati da molteplici fattori nella fase di produzione e confezionamento e portare a varie problematiche. Si tratta di difetti che possono portare all’inefficacia della terapia o, nel peggiore dei casi, ad effetti collaterali anche gravi.
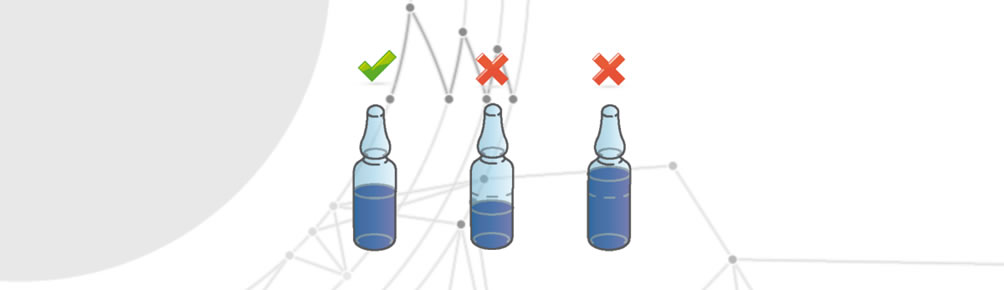
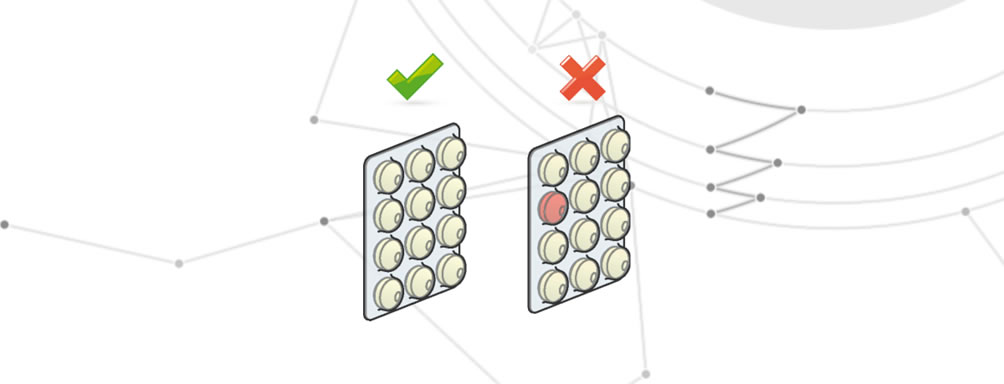
Quantità di farmaco non corretta
Si verifica quando una confezione monouso contiene una quantità di farmaco in eccesso o in difetto rispetto ai termini stabiliti.
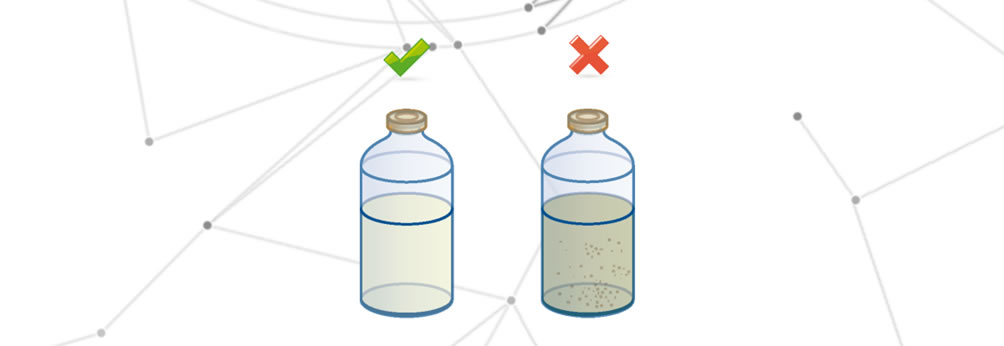
Degradazione del farmaco
Si parla di degradazione del farmaco quando si verifica una delle seguenti alterazioni:
- Modifica delle proprietà organolettiche.
- Totale o parziale riduzione dell’effetto terapeutico.
- Alterazione degli effetti sul paziente finale.
Cross contamination
Il difetto Cross contamination si verifica quando il farmaco presenta differenti principi attivi oltre a quello che dovrebbe contenere. Questi differenti principi attivi superano le tolleranze di accettabilità decise per legge. Un difetto che può derivare, ad esempio, da un processo di pulizia non corretto della linea di produzione tra un farmaco e un altro, oppure dalla presenza di agenti esterni.
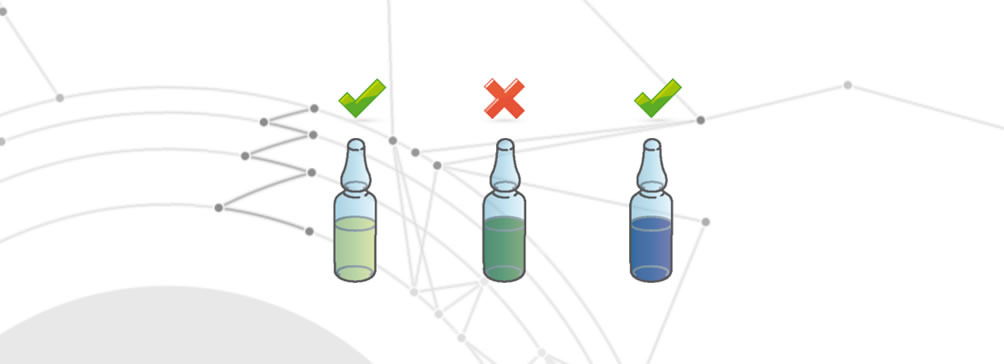
Prodotto errato
Per prodotto errato si intende l’uso di un prodotto che non è contemplato dal lotto di produzione.
GMP nel settore farmaceutico: Come individuare i difetti – Seconda parte
Foto di Anna Shvets da Pexels