LA VALIDATION PHARMACEUTIQUE
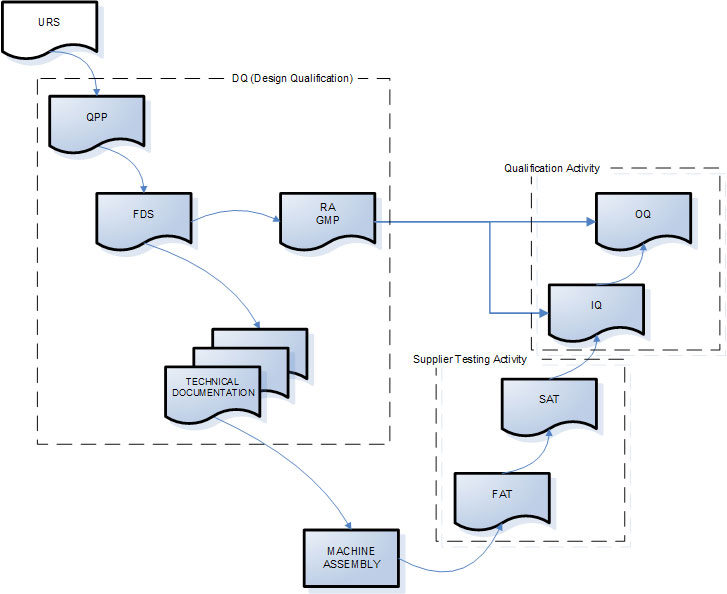
Afin que les compagnies pharmaceutiques peuvent produire ou confectionner les médicaments,les autorités concernées doivent délivrer une autorisation.l’autorisation est obtenue par l’usine s’il est possible de montrer que les processus de production et de l’emballage soient réalisées selon GMP (Good Manufacturing Practice).la démonstration de l’utilisation des processus qui suivent tels critères est obtenue par l’activité de la validation.la validation est une activité formelle qui suit tout le cycle de vie des processus, considérant en eux les machines, les structures, les méthodes, les usagers, les contrôles et en générale tout ce qui peut entraîner un danger pour l’utilisateur final, ou le patient.
Puisque la validation est une activité formelle, est nécessaire une évidence documentée des décisions prises et des activitées effectuées.l’évidence documentée est réalisée grâce à l’utilisation des documents rédigés, vérifiés et approuvés avant l’utilisation du personnel qui assume la responsabilité pour pouvoir décréter l’authenticité et l’utilisabilité.
QU’EST-CE QU’IL EST NÉCESSAIRE DE VALIDER?
D’une manière générale est nécessaire valider chaque aspect qui puisse avoir un impact sur la “ sécurité du patient “ en considérant que celle-ci vienne compromise de la perte d’intégrité des données critiques enregistrés, de la manque et/ou l’inexactitude des informations concernant l’usage du médicament et en générale de chaque aspect qui aille peser sur les attribués physiques, chimiques et les thérapeutiques du médicament .
LES PROCESSUS DE PRODUCTION ET DE L’EMBALLAGE
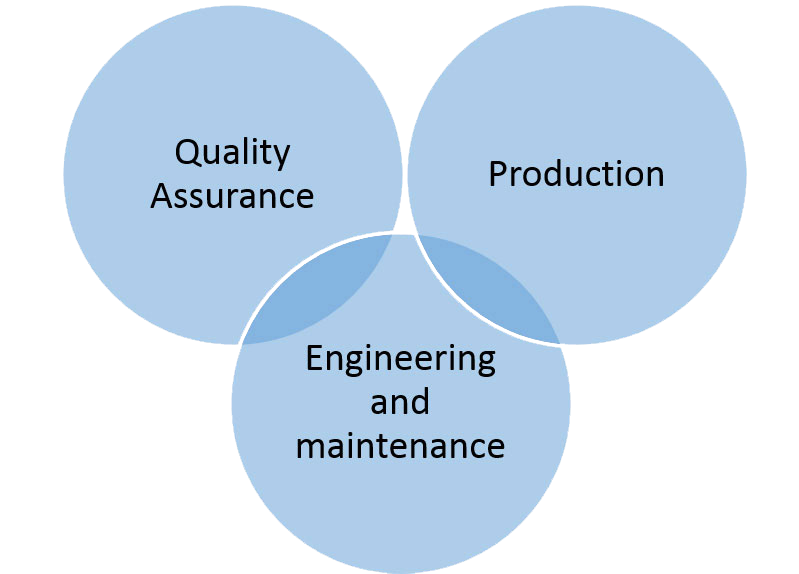
les médicament sont réalisés par des processus de production et d’emballage. Chaque processus est définie par l’ensemble des éléments suivants :