PHARMAZEUTISCHE VALIDIERUNG
Damit Pharmaunternehmen Arzneimittel herstellen oder verpacken können, müssen die zuständigen Behörden eine Zulassung erteilen. Das Unternehmen erhält die Zulassung wenn es nachgewiesen werden kann, dass die Produktions- und Verpackungsprozesse nach GMP (Good Manufacturing Practice) durchgeführt werden. Durch die Validierung wird es nachgewiesen, dass die Prozessen alle notwendigen Kriterien erfüllen. Die Validierung ist eine formale Aktivität, die den gesamten Lebenszyklus der Prozesse kontrolliert, und zwar die Maschinen, Strukturen, Methoden, Kontrollen und im Allgemeinen alles berücksichtigt, was eine Gefahr für den Endbenutzer, nämlich den Patienten, darstellen kann.
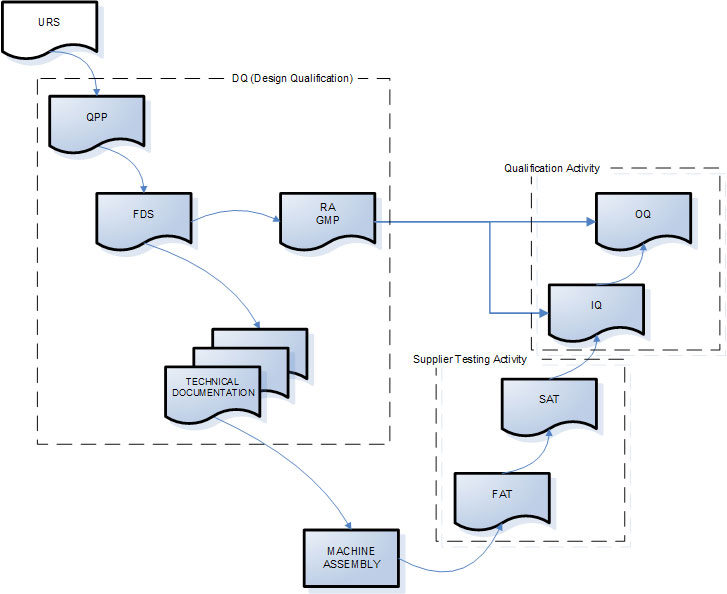
Da es sich bei der Validierung um eine formelle Tätigkeit handelt, ist ein dokumentierter Nachweis der getroffenen Entscheidungen und durchgeführten Tätigkeiten erforderlich. Der dokumentierte Nachweis erfolgt durch die Verwendung von Dokumenten, die vor der Verwendung von dem verantwortlichen Personal erstellt, geprüft und genehmigt werden, um deren Echtheitund Verwendbarkeitzu erklären.
WAS SOLL VALIDIERT WERDEN?
Im Allgemeinen ist es notwendig, jeden Aspekt zu validieren, der sich auf die „Patientenssicherheit“ auswirken kann. Die Sicherheit der Patienten kann durch den Verlust der Integrität der gesammelten kritischen Daten, durch das Fehlen und/oder die Unrichtigkeit von Informationen in Bezug auf die Verwendung des Medikaments und im Allgemeinen durch jeden Aspekt, der die physikalischen, chemischen und therapeutischen Eigenschaften des Medikaments betrifft, beeinträchtigt werden.
DER PRODUKTIONS- UND VERPACKUNGSPROZESS
Die Medikamente werden durch Herstellungs- und Verpackungsprozessen hergestellt. Jeder Prozess wird anhand von folgenden Kriterien definiert: